




The Process of Drug Development: An Overview
Taking new drugs, treatments, or therapies to market is a demanding, expensive, and time-consuming journey from an initial idea to a product ready to launch in the marketplace. In some cases, the research and development (R&D) process for new treatments and therapies can take more than a decade and cost billions of dollars.
Following is an overview of the drug discovery and development process typically used to bring new therapies to market.
The importance of drug discovery and development
Pharmaceutical companies spend up to $4.5 billion for each new treatment they work to bring to market. In terms of time, the entire process can take more than 13.5 years to complete.
Some estimates say that up to two-thirds of the total time spent on R&D comes during the drug development process when researchers work to establish the safety and benefits of potential new therapies.
The U.S. Food and Drug Administration (FDA) identifies five steps in the FDA drug development process.
They are:
- Discovery and development. Discovery is when research scientists discover new therapies and identify which look most promising and deserve additional study. Those therapies are then studied further to determine their efficacy, benefits, possible side effects, and other information.
- Preclinical research. Potential treatments are tested in laboratories and animals to help answer basic questions about safety and if they should be tested on people.
- Clinical research. If preclinical testing indicates the therapies are safe, they then move into clinical testing on people to make sure they are effective and safe.
- FDA review. Review teams from the FDA examine the results of clinical trials and other research to decide if they will approve or not approve the new therapy.
- FDA post-market safety monitoring. The FDA continues to monitor all approved treatments and therapies for safety after they have been approved for use by the public.
The journey of a new therapy
There are many steps in the journey of a potential new treatment or therapy as it moves from initial discovery to development, through preclinical and clinical trials, and finally to market.
From the initial scientific insight that starts the research and discovery process through the final product launch, there are many steps potential treatments must complete to prove their safety and efficacy.
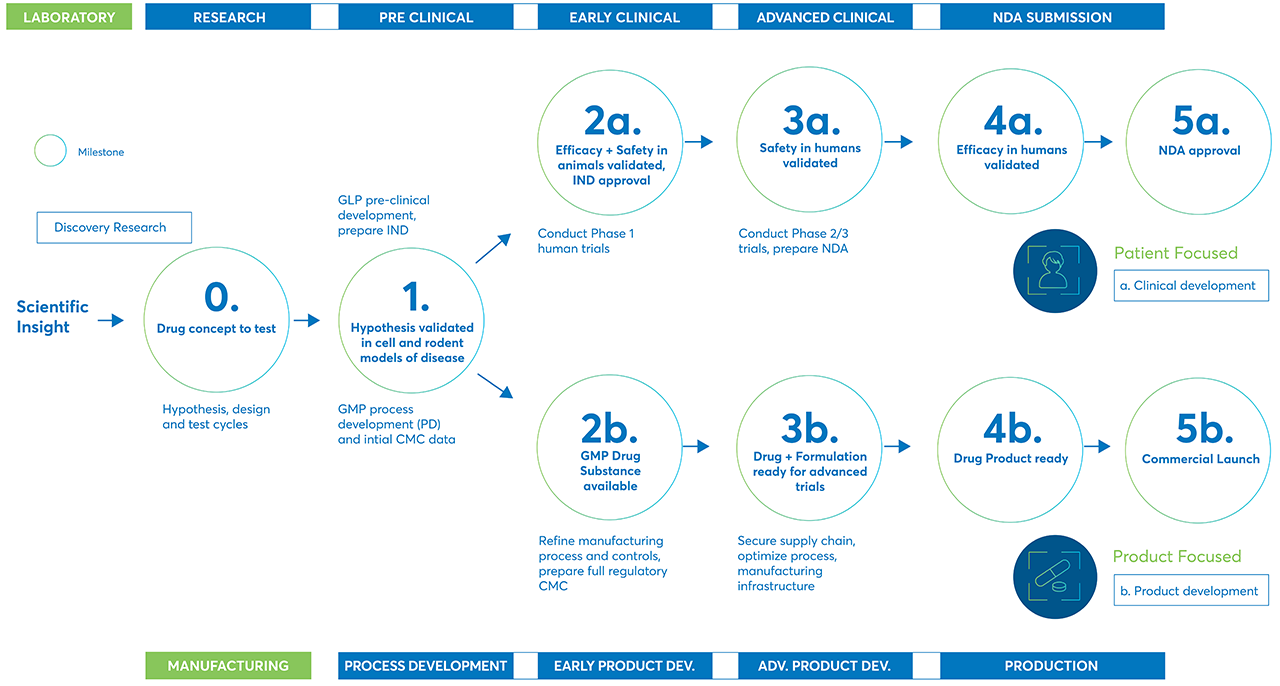
The process starts with discovery
Discovery is the first step in bringing new treatments and therapies to market. In the process stage, researchers evaluate compounds to determine which could be candidates for development as medical treatments.
A key goal for the discovery phase is to identify new molecules and chemical compounds that could be effective therapy for treating diseases or be more effective than existing therapies. Researchers use several methods in the discovery process to identify and evaluate potential treatments. They include:
- Testing many molecular compounds to find if they offer possible benefits against any of many diseases
- Retesting existing treatments to see if they offer benefits against other diseases
- Using new information about diseases to help design products that could stop or reverse the effects of the disease
- Adopting new technologies to treat diseases, such as those that provide the ability to target specific sites within the body or manipulate genetic material
During the process's discovery stage, researchers may look at thousands of compounds to identify candidates for development as a therapeutic treatment. Often, only a few compounds look promising and call for further study.
The Pharmaceutical Journal, a journal based in the United Kingdom, estimates that for every 10,000 compounds tested in the discovery stage, only 10 to 20 compounds move on to the development phase. Only about half of those that enter the development proceed into preclinical trials.
Drug development is the next step
After researchers identify a promising compound during the discovery phase, the compound moves into the development phase. In that phase, scientists conduct a series of experiments on the compound to gather information on:
- How the therapy is absorbed, distributed, metabolized, and excreted
- The potential benefits of the therapy and how it works
- The best dosage for the proposed treatment
- The best way to administer the therapy, such as orally or by injection
- Any side effects or adverse events that could be considered toxicity
- If the therapy affects specific groups of people—such as gender, race, age, or ethnicity—differently from other groups
- How the new therapy interacts with other drugs and treatments
- The therapy’s effectiveness compared to existing treatments
The drug development phase includes everything that must be completed to transform a compound from a therapy candidate at the end of the discovery phase to a product approved for marketing by the appropriate regulatory authorities.
Speed and efficiency during this process can be critical for the commercial success of new treatments and therapies. One reason is that the time spent in the development process counts against its patent protection period after the drug goes to market. When the patent expires, generic competition can sharply reduce sales revenue.
Another reason cost control is a significant concern is that if the compound does not successfully make it through the development process and approval stages, the process must begin again, and the time, money, and resources invested may be lost. However, many biomanufacturers learn a great deal through failures, and in many cases, the lessons they learn drive subsequent breakthroughs and innovations.
Preclinical research
Potential therapies cannot be tested on humans until researchers have completed preclinical research showing that the potential treatment is not likely to cause serious harm, called toxicity, when used in humans. There are two types of preclinical research:
- In vitro
- In vivo
In vitro experiments are performed under controlled laboratory conditions outside a living organism. In contrast, in vivo experiments are physiological research performed inside a living organism, such as mice or other animals.
To help ensure the consistency of results, the FDA requires that researchers use good laboratory practices (GLP) for preclinical laboratory studies. The GLP regulations set minimum basic requirements for:
- Study conduct
- Personnel involved
- Facilities
- Equipment
- Written protocols
- Operating procedures
- Reports
The GLP also specifies a system of quality assurance and oversight for each study to help ensure the safety of FDA-regulated products.
Though preclinical studies are usually not extensive, the results must provide detailed information on dosing and toxicity levels. After preclinical testing, researchers review the results to determine if the treatment should move to the clinical research step for testing in people.
Clinical Research
Clinical trials are research that involves people. After preclinical research has answered basic questions about a therapy’s safety, it is time to move to clinical trials, the stage when researchers can gather more information about safety and effectiveness by studying the drug as it is used in humans.
Clinical trials should be designed to answer specific questions about a treatment or therapy. The trials are conducted according to a specific plan, called a protocol, developed by researchers or manufacturers. Researchers review existing information about the treatment as they develop the protocol to establish their research questions and objectives. Based on those, they then decide:
- The selection criteria, or who qualifies to participate in the trial
- The number of people who will be part of the trial
- The length of the study If there will be a control group or other ways to limit research bias
- The dosage and how the drug will be administered
- When and how data will be collected and assessed
- How data will be reviewed and analyzed
Clinical trials are typically conducted in three phases, ranging from small-scale Phase 1 studies to large-scale Phase 3 trials. If a therapy performs well in one phase, it moves on to the next phase for additional research.
- Phase 1 trials usually involve less the 30 people and are designed to find a safe dose and the best way to administer the therapy. Phase 1 trials also help make sure the treatment is safe for the human body. Phase 1 trials usually last several months. About 70% of drugs move on to the next phase.
- Phase 2 trials include up to 100 patients and study how the treatment affects a condition or disease and the human body. Phase 2 trials can take up to two years to complete. Approximately one-third of drugs in Phase 2 move on to the next phase.
- Phase 3 trials include anywhere from 100 to several thousand participants. In Phase 3 trials, the new therapy is compared to existing treatments to see which is more effective. Phase 3 trials can take one to four years to complete. Between 25 and 30% of new drugs complete Phase 3 trials.
The FDA review process
Clinical trial sponsors must submit an Investigational New Drug (IND) application before testing a new therapy on human participants. When a sponsor submits an IND, the FDA assembles a review team with the following scientific specialists, each with different responsibilities.
- Project Manager. The project manager coordinates the team’s activities throughout the review process and serves as the primary contact for the trial’s sponsor.
- Medical Officer. The medical officer reviews clinical trial data before, during, and after its completion.
- Statistician. The statistician works closely with the medical officer to evaluate protocols and review safety and efficacy data.
- Pharmacologist. The pharmacologist reviews preclinical studies to ensure the therapy is safe to proceed into clinical trials.
- Pharmakineticist. Pharmakineticist focuses on the drug’s absorption, distribution, metabolism, and excretion processes.They also review blood-level data at different intervals during the trial to assess dosages and administration schedules.
- Chemist. The chemist evaluates a drug’s chemical compounds and analyzes how it was made, its stability, quality control, continuity, the presence of impurities, etc.’
- Microbiologist. The microbiologist reviews submitted data for antimicrobial products to assess how well they work with different classes of microbes.
FDA IND reviews protect volunteers participating in clinical trials from unreasonable or significant risks. The FDA review team has 30 days to review an IND submission. It can either grant approval to begin a clinical trial or issue a clinical hold to delay or stop the investigation. The FDA can place a clinical hold for several reasons, including:
- Participants could be exposed to unreasonable or significant risk.
- The investigators are not qualified.
- The informational materials for volunteer participants are misleading.
- The IND application does not include enough information about risks.
In most cases, the FDA approves the clinical trial if the proposed study meets federal standards.
Researchers and sponsors are responsible for informing the review team about any new protocols or severe side effects during the trial so that the team can monitor trials carefully for signs of problems.
This process continues until the developer ends clinical trials or files a marketing application. Before filing a marketing application, a developer must have adequate data from two large, controlled clinical trials and submit study reports.
FDA reviews and post-market monitoring
When trials are complete and the data submitted to the FDA for approval, The regulatory authority is responsible for the scientific evaluation of the NDA or MAA. The goal of the application is to provide regulators with enough information – gathered during preclinical and clinical studies – for them to be able to determine if:
- The therapy is safe and effective for the condition it was developed to treat
- The treatments benefits outweigh any risks
- The therapy’s labeling is fit-for-purpose and includes all required details
- Manufacturing methods and quality control are satisfactory
The FDA continues to monitor a therapy’s safety and efficacy after it has been approved and taken to market. That’s because no matter how extensive clinical trials were, complete information about a therapy’s safety and efficacy may not be known for months or years of use.
The FDA reviews reports of problems with an approved therapy through post-market drug safety monitoring. Depending on the severity of the problem, the FDA could add caution statements to the dosage or usage information. Or it could take other measures for more serious issues.
There are other instances in which additional trials or research may be required. They include:
Supplemental applications. If a developer wants to make changes in formulation, labeling, or dosage strength, it must file a supplemental application and the change approved by FD.
INDs for marketed drugs. Developers must file a new IND if they want to further develop an approved drug for a new use, dosage strength, new or different form—such as an injectable or oral liquid instead of a tablet—or if they want to conduct other clinical research or a post-market safety study.
Avantor expertise can drive your clinical trials forward
Avantor® Clinical Services can help you maximize clinical trial efficiency by providing scalable, end-to-end service capabilities that can help streamline the clinical trial lifecycle. Our services let you focus on your research and patient outcomes.
We offer a full range of clinical trial services that cover every step, from discovery to delivery. Our portfolio of custom kitting, equipment and ancillary solutions, and biorepository and archiving services is ready to help you with your trial.